Jatketaan tauon jälkeen blogin päivittämistä niinkuin mitään ei olisi tapahtunut!
Edellisissä postauksessa käsittelimme luonnonvalintaa yhden lokuksen näkökulmasta. Moniin fenotyyppisiin ominaisuuksiin vaikuttavat kuitenkin monet geenit, sekä ympäristö. Tietysti voisimme pitää kirjaa kaikista lokuksista erikseen, mutta lokusten määrän kasvaessa ja varsinkin jos eri lokukset ovat fyysisesti lähellä toisiaan ja eivät periydy toisistaan riippumatta, tilanteesta tulee nopeasti melko monimutkainen. Lisäksi emme useinkaan tiedä kaikkia johonkin ominaisuuteen vaikuttavia geenejä. Niinpä olisikin hyvä jos voisimme tarkastella luonnonvalinnan toimintaa pelkän fenotyypin tasolla.
Iloksemme voin todeta, että tämä on erittäin toimiva ja hyödyllinen lähestymistapa. Tosin ennen kuin voimme käyttää tätä tapaa, meidän täytyy tehdä joitakin oletuksia joiden oikeutuksesta lienee syytä ensin keskustella.
Opimme aiemmin Mendelin lait ja niitä tarkasteltaessa tulimme siihen tulokseen, että perinnöllisyys on diskreettiä. Jos näin on, miten voimme selittää monet havainnot, joiden mukaan luonnossa esiintyvä muuntelu fenotyypeissä näyttäisi olevan jatkuvaa eikä sitä voida jakaa erilaisiin luokkiin. Esimerkiksi ihmisten pituus näyttää noudattavan normaalijakaumaa (kellokäyrä). Mendelin tutkimat ominaisuudet näyttävät olevan enemmän poikkeus kuin yleinen asioidentila. Tämä kysymys askarrutti monia luonnontutkijoita, kun Mendelin tulokset tulivat yleiseen tietoon. Monet aikalaiset olivatkin sitä mieltä, että Mendelin lait eivät voineet selittää perinnöllisyyttä ja evoluutiota.
Mendelin lakien ja havaintojen yhteensopimattomuuden ongelman ratkaisi R. A. Fisher vuonna 1918 artikkelissaan: ”The correlation between relatives on the supposition of Mendelian inheritance”. Siinä Fisher osoitti, että jos populaatiossa on useita lokuksia, joiden eri alleelit vaikuttavat fenotyyppiin eri tavoin, fenotyyppinen muuntelu populaation tasolla lähestyy normaalijakaumaa, vaikka kaikki yksittäiset lokukset noudattavat Mendelin lakeja. Niinpä ristiriita Mendelin havaintojen ja luonnossa esiintyvän muuntelun kesken ei ole todellinen.
Kvantitatiivisen genetiikan perusmalli
Voimme havainnollistaa kvantitatiivisen genetiikan perusteita seuraavasti: olettakaamme että tarkastelemme fenotyyppistä ominaisuutta 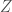
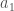


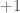

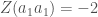

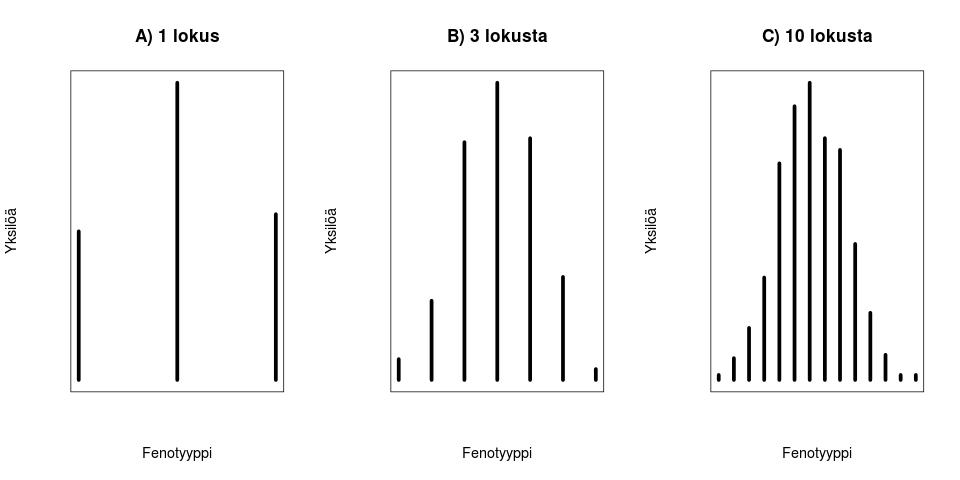
Fenotyyppi on jakautunut diskreetisti populaatiossa. Kun lisäämme lokusten määrää joissa esiintyy geneettistä muuntelua (olen tässä esimerkissä skaalannut myös yksittäisten lokusten vaikutuksia, jotta voimme vertailla jakaumia helposti) huomaamme, että fenotyypin jakauma lähestyy normaalijakaumaa (Kuva 1, B ja C) kun lokusten määrä jossa on on fenottyyppiin vaikuttavaa geneettistä muuntelua kasvaa. Luonnossa useimpiin mitattaviin ominaisuuksiin vaikuttaa tietysti myös satunnaisvaihtelu joka voi olla seurausta siitä, että otukset ovat kehittyneet erilaisissa ympäristöissä tai satunnaisesta vaihtelusta yksilönkehityksen aikana. Voimmekin kuvata jonkin yksilön 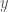
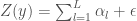


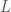

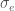
Jos oletamme, että mielenkiinnon kohteena olevaan ominaisuuteen vaikuttaa useita lokuksia sekä ympäristön aikaansaama satunnaisvaihtelu, mutta emme tunne kaikkia ominaisuuten vaikuttavia lokuksia. Myöhemmin tulemme huomaamaan, että kun lokuksia on monia, niiden tilastollinen käyttäytyminen on sellaista, että voimme käyttää tilastollisia menetelmiä esim. luonnonvalinnan tutkimiseen, vaikka emme tunnekaan yksittäisten ominaisuuten vaikuttavien lokusten genotyyppejä. Tässä on hieman sama idea kuin fysiikassa tutkittaessa esim. ideaalikaasun tms. käyttäytymistä: vaikka emme tiedä yhdenkään molekyylin tarkkaa paikkaa tietyllä ajanhetkellä, voimme silti sanoa kaasun käyttäytymisestä jotain. Tästä genetiikan haarasta käytetään nimeä kvantitatiivinengenetiikka (joskus näkee puhuttavan myös tilastollisesta genetiikasta hieman laajemmassa merkityksessä). Näitä tekniikoita käytetään sellaisessa kasvi- ja eläinjalostuksessa, joka perustuu valintaan (valtaisa enemmistö kaikesta jalostustoiminnasta). Lisäksi tekniikat ovat tärkeitä evoluutiotutkimuksessa.
Mallin oletuksista
Ensimmäinen kysymys joka tietenkin herää on se, että onko useimpien fenotyyppien muunteluun osallisena monia lokuksia niin kuin teoria edellyttää? Modernein menetelmin on pystytty selvittämään, että näin todella on. Alunperin Fisher ajatteli, että jokaiseen ominaisuuten vaikutti lukematon määrä lokuksia. Tällä hetkellä näyttäisi siltä, että määrä on rajallisempi. Lisäksi ilmeisesti usein on niin, että yksittäisten lokusten vaikutukset eivät jakaannu tasaisesti vaan muutamilla lokuksilla voi olla (suhteellisesti) suuriakin vaikutuksia, mutta lokuksia joilla on pieniä tai erittäin pieniä vaikutuksia on paljon enemmän. Niinkuin edellä todettiin, tämä ei kuitenkaan ole oletuksien kannalta ongelma.
Hyödyllisyydestä
Mitä suuri yleisö ei välttämättä tiedosta on se, että vaikka seuraavaksi esitettävät tilastolliset menetelmät saattavat joistakin lukijoita tuntua hieman vanhanaikaisilta (ovathan ne suurimmaksi osin jo hyvin vanhoja, koska perusteet kehetettiin jo 1900-luvun alussa) ne toimivat vieläkin monissa tapauksissa paremmin kuin moderneiksi mielletyt menetelmät. Esimerkiksi, jos minun pitäisi ennustaa blogin lukijan pituus (Esitän kysymyksen tässä tietysti pointin selittämisen vuoksi, oikeasti helpointa ja tarkinta olisi tietysti kysyä tai mitata itse.), niin tarkemman ennusteen saisi mittaamalla lukijan isän ja äidin pituudet kuin sekvensoimalla lukijan koko genomin. Tiedämme kuitenkin vasta niin vähän monien ominaisuuksien geneettisestä taustasta. Tässä ongelmana on että niiden lokusten, joilla on vain vähäinen vaikutus fenotyyppiin, havaitseminen empiirisessä tutkimuksessa on vaikeaa. Niinpä niillä geeneillä joiden tiedetään vaikuttavan johonkin kvantitatiiviseen ominaisuuteen, on usein suhteellisesti suuri vaikutus.
Ennen kuin lähdemme tarkastelemaan, miten voimme tutkia luonnonvalintaa kvantitatiivisen genetiikan menetelmin on syytä muistuttaa muutamasta seikasta. Aina kun tarkastelemme johonkin ominaisuuten vaikuttavia lokuksia tai geenejä, tarkoitamme tällä lokuksia joissa on geneettistä muuntelua, eli alleeleja joilla on erilaiset vaikutukset tarkasteltavaan fenotyyppiseen ominaisuuteen. Kaikki sellaiset geenit, joissa ei esiinny muuntelua, mutta jotka ovat jonkin ominaisuuden kannalta tärkeitä eivät tietenkään näy populaation fenotyyppisessä muuntelussa. Tähän palaamme myöhemmin.
Kirjallisuutta
Fisher, R. A. 1918. The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh. 52: 399-433
